Mechanisms of prion replication, spread and toxicity
Current State of Prion Science
Prions are composed of PrPSc, infectious aggregates (Prusiner, Science 1982) of the cellular prion protein (PrPC, encoded by the PRNP gene) that progressively accumulate in the infected brain.
Prions cause neuropathological damage (astrogliosis, excessive microglial activation, neuronal loss, amyloid plaques) (Aguzzi and Polymenidou, Cell 2004) and spongiosis resulting from neuronal vacuolation. Nucleation and self-sustained propagation of protein aggregates, once thought to be limited to prion diseases, is now recognized as an important principle in many disorders (Aguzzi, Nature 2009; Science 2024). The incidence of these diseases is rising, and disease-modifying therapies are lacking. This bleak situation is compounded by a dearth of validated therapeutic targets, which stems from our incomplete understanding of the biochemical and cellular networks involved in the formation, propagation, clearance, and toxicity of prions (Aguzzi et al., Annu Rev Pharmacol Toxicol 2018). In the past decades, human genetics revealed the existence of important risk genes specifically associated with neurodegenerative pathologies.
The quest for prion-related risk factors was more challenging, as all the genetic forms of prion disease are caused exclusively by mutations of the PRNP gene. The onset of prion diseases caused by PRNP mutations (e.g. E200K) ranges between 31 and 92 years (Minikel et al., Neurology 2019), pointing to the existence of strong disease modifiers. Yet genome-wide association studies have only highlighted STX6 and GAL3ST1 as weakly associated with sporadic prion disease (Jones et al., Lancet Neurol 2020; Jones et al., Neurobiol Dis 2024; Sangar et al., eLife 2024; Hill et al., bioRxiv 2025). Crucially, the generation of prion infectivity ab initio from recombinant PrPC (Legname et al., Science 2004) requires unphysiological perturbations such as shearing or denaturation, whereas administration of infectious prions to susceptible cells leads to immediate and efficient prion replication. This suggests the existence of a hitherto undiscovered "prion synthase" capable of multiplying prions in cellula.
Ongoing efforts at ISAB.
Targeted whole-genome perturbation is revolutionizing our understanding of complex biological systems. At ISAB, we apply arrayed and pooled CRISPR-based gain- and loss-of-function approaches to systematically identify genes and pathways that modulate the formation, toxicity, clearance, and spread of prions.
Recent evidence indicates that most human amyloidoses, including highly prevalent proteinopathies of the brain and other organs, follow principles of seeded aggregation and prion-like propagation. By combining functional genomics with biochemical, cell-biological, and in vivo approaches, ISAB aims to uncover conserved and disease- specific mechanisms that govern pathological protein aggregation across neurodegenerative disorders.
Contribution of the Aguzzi lab to prion science
- Co-discovered (with C. Weissmann) that removal of Prnp protects from prions ( Bueler et al., Cell 1993 ).
- Identified PrPC as a mediator of prion toxicity ( Brandner et al., Nature 1996 ).
- B-lymphocytes are necessary for prion neuroinvasion ( Blättler et al., Nature 1997 ; Klein et al., Nature 1997 ).
- Showed that plasminogen is the carrier protein of prions in blood ( Fischer et al., Nature 2000 ).
- Showed that antiprion antibodies are protective against prions in vivo ( Heppner et al., Science 2001 ).
- Showed that immunoadhesins are protective against prions in vivo ( Meier et al., Cell 2003 ).
- Clarified the mechanism of prion entry into peripheral nerves ( NEJM 2003 ; Glatzel et al., Neuron 2001 ).
- Showed that inflammation controls the organ tropism of prions ( Ligios et al., Nat Med 2005 ; Seeger et al., Science 2005 ).
- Showed that PrP is essential for axomyelinic integrity ( Bremer et al., Nat Neurosci 2010 ).
- Follicular dendritic cells arise from pericytes and produce prions in lymphatic tissue ( Krautler et al., Cell 2012 ).
- Identified neurotoxic and neuroprotective domains of the prion protein ( Sonati et al., Nature 2013 ).
- Identified polythiophenes as antiprion compounds ( Herrmann et al., Sci Transl Med 2015 ).
- Identified Adgrg6 as a mediator of prion protein function ( Kuffer et al., Nature 2016 ).
- Found natural protective antiprion antibodies in humans ( Senatore et al., EMBO Mol Med 2020 ).
- A PrPC conformational switch triggers prion toxicity ( Frontzek et al., Nat Struct Mol Biol 2022 ).
- Identified hnRNP K as a limiting factor for prion replication ( Avar et al., EMBO J 2022 ).
- Identified K27 as a residue mediating toxicity in the N-terminus of PrP ( Reimann et al., Brain Pathol 2023 ).
- Discovered a role of NG2 glia in protecting against prion neurotoxicity ( Liu et al., Nat Neurosci 2024 ).
- Produced the first arrayed genome-wide CRISPR lentiviral libraries to study prion diseases ( Yin et al., Nat Biomed Eng 2024 ).
Outlook
We argue that the therapy of neurodegeneration is held back by a dearth of actionable therapeutic targets. We have developed a rich toolbox with which we will address this question.
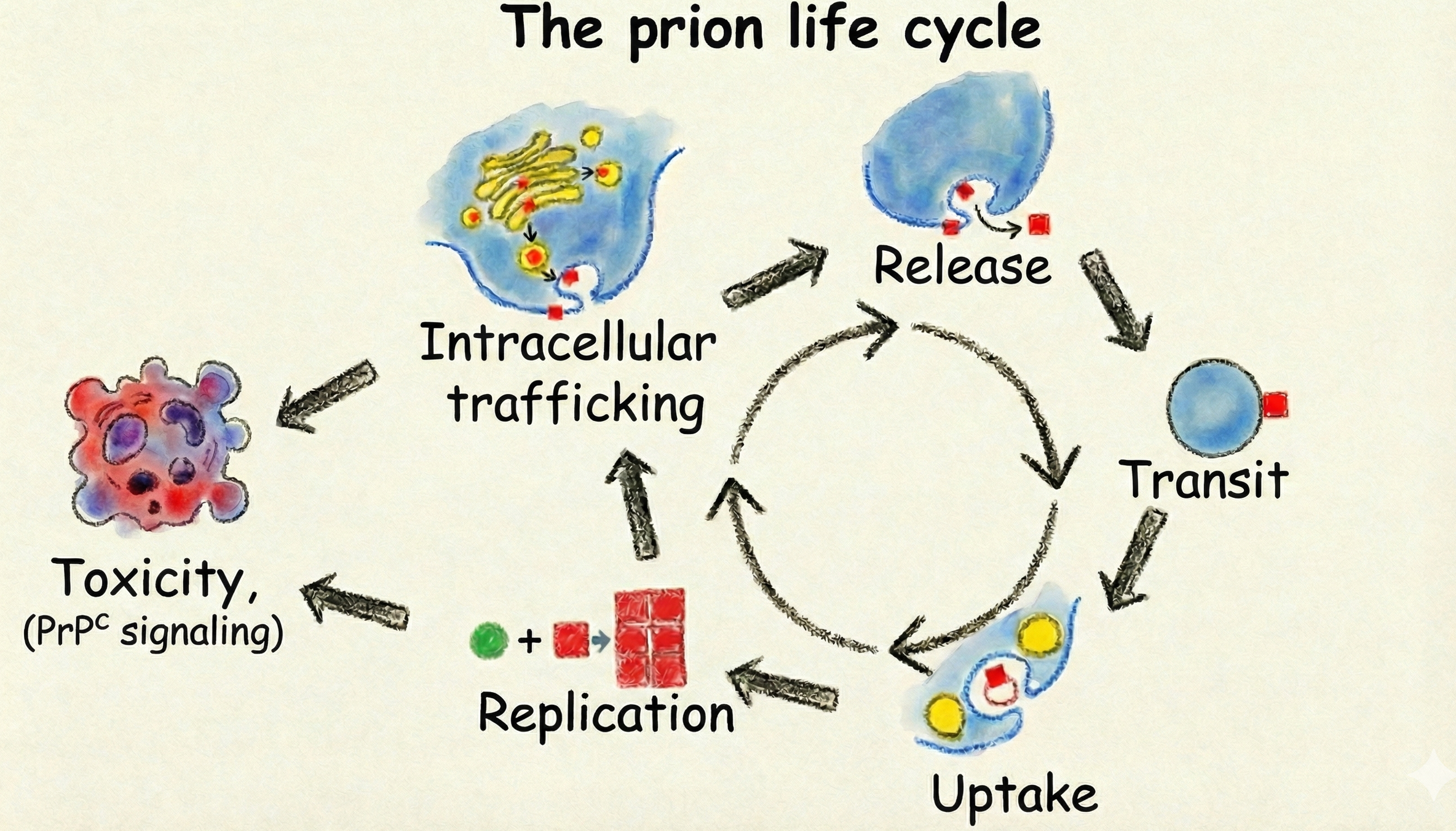
Objective #1 is centered on how prions propagate in the brain of infected individuals. Prion propagation proceeds through three consecutive steps: internalization, replication and transcellular transfer. The 1st step is obviously crucial, yet no prion-specific receptors are known. We have identified potential modulators of prion internalization and cell-to-cell transfer, which we will intersect with the data from our previous screens and from human genetics. The most promising hits will be characterized mechanistically. Prion replication and transfer will be investigated through a comprehensive, multi-tiered screening approach utilizing a co-culture system of prion-infected and naïve cells. Targeted biochemical assays will subsequently differentiate between hits that influence transcellular transfer and genes acting on the replication process.
Objective #2 will explore the cellular and molecular basis of prion toxicity. Synthetic lethality screens performed in our lab have identified ciliary assembly/disassembly as potent modifiers of prion toxicity. Excitingly, their pharmacological targeting appears to impact disease progression. The effect of ciliogenesis-related pathways will be assessed in models of prion disease, and novel transduction pathways involved in prion toxicity cascades will be investigated.
Relevance and Impact
Although prion research was long considered a niche area, the past decade has shown that prion-like phenomena are pervasive in all life forms, controlling both physiological states as "functional amyloids" (Balistreri et al. Microorganisms 2020) (Aguzzi and Rajendran Neuron 2009) and pathological conditions. Although the pace of prion-related discoveries has experienced a golden age of rapid progress in 1980-2000 (Aguzzi Science 2024), it has often stagnated in recent years (Aguzzi and De Cecco Science 2020). This proposal aims at breaking this impasse. According to Thomas Kuhn, such impasses are often overcome when a new methodology engenders a paradigm shift. The present proposal is based on the premise that CRISPR-based arrayed forward genetics screens represent such a disruptive technology (Aguzzi and Kampmann Science 2023). Our preliminary results indicate that unbiased, hypothesis-free experimentation, in conjunction with sophisticated methods of functional genomics, can indeed identify molecules and pathways of prion replication, spread and toxicity that would not be discoverable by any other approach. Once the "partners in crime" of PrPC are identified, their role will be tested using orthogonal methods and technologies. One may criticize that the proposal is too broad and ambitious. However, the technologies required are already established in our lab, and the technical risks are contained. Finally, each objective is addressed from several angles, thereby providing contingency plans if any of the proposed approaches meet with difficulties.
This project will have a far-reaching impact on the academic community by advancing both research and education in neurodegeneration, molecular genetics, and prion biology. The integration of cutting-edge genomic screening techniques and advanced imaging approaches will provide an invaluable framework for training the next generation of scientists. The project will generate novel datasets and methodological advancements that can be incorporated into university curricula, graduate courses, and specialized workshops, enriching educational programs in neuroscience and molecular biology. Additionally, the project's findings will be widely disseminated through publications, conferences, and interdisciplinary collaborations, fostering knowledge exchange and inspiring future research on protein misfolding disorders. By bridging fundamental science with translational potential, this work will contribute to a deeper understanding of neurodegenerative diseases while strengthening academic engagement in the field.